An unusual ulcer in an 8-year-old girl
Published Web Location
https://doi.org/10.5070/D36wn5b7swMain Content
An unusual ulcer in an 8-year-old girl
Adam Friedman MD1, Maria Abadi MD2, Katherine Wu3, Michael Fisher1, Jacob Abadi MD4,5
Dermatology Online Journal 16 (2): 6
1. Division of Dermatology, Department of Medicine, Albert Einstein College of Medicine, Bronx, New York. ajf0424@yahoo.com2. Department of Pathology, Albert Einstein College of Medicine Bronx, New York
3. Albert Einstein College of Medicine, Bronx, New York
4. Department of Pediatrics, Albert Einstein College of Medicine, Bronx, New York
5. Division of Infectious Diseases, Department of Pediatrics, Albert Einstein College of Medicine, Bronx, New York
Case presentation
An 8-year-old girl from the Dominican Republic with no significant past medical history presented with a 4-week history of fever and a progressively enlarging painless ulcer with adjacent violaceous plaques on the left leg. Six weeks prior to admission, the patient fell sustaining trauma to the posterior thigh with the edge of a concrete step. The resulting injury resolved with nonsteroidal anti-inflammatory therapy. However, cutaneous findings returned two weeks later with concomitant fever and rapid progression. Treatment in the Dominican Republic with IV Penicillin was ineffective.
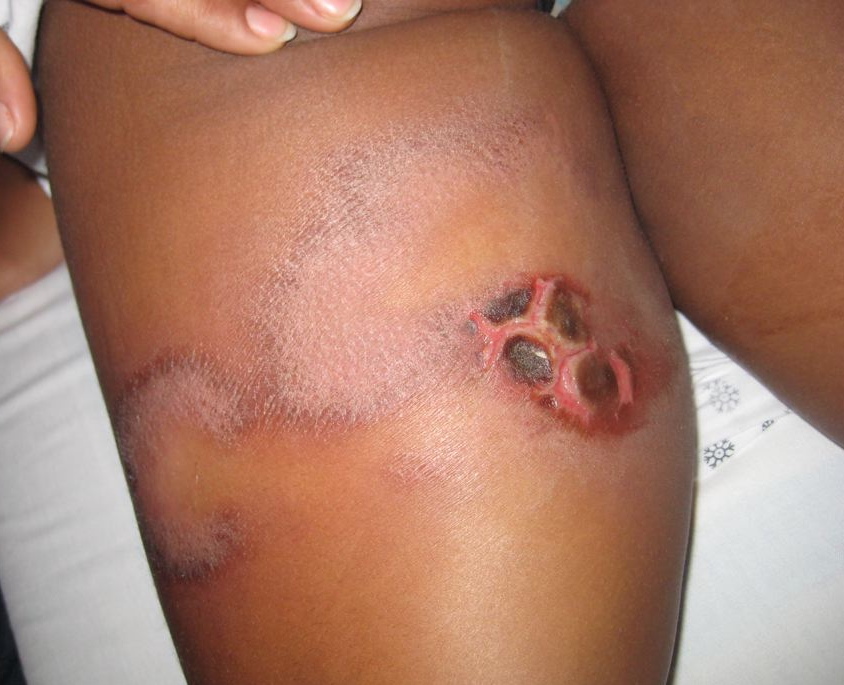 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Ulcer with islands of eschar on posterior left leg with associated indurated, erythematous semi-annular plaques Figure 2. (A) An incisional biopsy showing a dense lobular panniculitis with septal fibrosis (H&E, x100). (B) Atypical lymphoid infiltrate circumscribing the adipocytes (H&E, x400) | |
Physical exam revealed a well-defined, non-underminable ulceration with islands of eschar on the left mediosuperior thigh (Figure 1). Laterally, there were two associated indurated, semi-annular plaques. Significant laboratory test results upon admission demonstrated pancytopenia. An incisional biopsy was performed and stained with hematoxylin-eosin (Figure 2) as well as with a panel of immunohistochemical markers. Samples were sent for cultures.
Histopathologic examination revealed an extensive atypical lymphoid infiltrate in the subcutaneous tissue with karyorrhexis, ulceration and necrosis. Immmunostains performed revealed neoplastic cells positive for CD2, CD3, CD5, CD7, CD8, granzyme B, and T cell intracellular antigen (TIA-1); negative for CD4 and CD56. Clonal T cell receptor-beta rearrangement was detected. Bone marrow biopsy failed to demonstrate atypical lymphoid aggregates, interstitial infiltration or hemophagocytosis. Tissue cultures showed no growth of bacterial, mycobacterial, or fungal species.
Subsequent bone marrow biopsy demonstrated severe hypocellular marrow with maturing trilineage hematopoiesis and patchy fibrinoid necrosis. There was no evidence of lymphoma or hemophagocytic syndrome. Flow cytometry did not demonstrate evidence of B cell clone or loss of T cell surface antigens.
The diagnosis of subcutaneous panniculitis-like T cell lymphoma was made and in collaboration with the division of Oncology, the patient was started on a chemotherapy regimen of cyclophosphamide, hydroxydaunomycin (doxorubicin), Oncovin (vincristine) and prednisone (CHOP). We fortunately report that she responded well to therapy and has been in remission for over one year.
Discussion
Subcutaneous panniculitis-like T cell lymphoma (SPTL) is not only a rare subgroup of cutaneous T cell lymphomas (CTCL), but it is somewhat of a misnomer - it is not a true panniculitis. Initially described in 1991 as an atypical presentation of T cell Lymphoma, SPTL is now recognized as a distinct subset of peripheral T cell lymphomas by the WHO [1], and incorporated in the classification of hematopoietic and lymphoid tumors [2].
SPTL frequently presents with tender, erythematous, dermal/subcutaneous nodules and plaques, often occurring on the extremities and trunk with approximately equal gender distribution. Median age at diagnosis is 36 years and median duration from cutaneous presentation to diagnosis is approximately 7 months [3]. Constitutional symptoms are seen in 40 percent of the cases, including fevers, weight loss, hepatosplenomegaly, and pancytopenia. Association with hemophagocytic syndrome (HPS) is seen in 45 percent of the cases [4] and carries a worse prognosis. Because cytophagic histiocytic panniculitis can also be associated with hemophagocytosis, a relationship between the two has been raised [5, 6]. These entities may be part of a clinical spectrum, demonstrating disease progression from a benign panniculitis to a malignant lymphoma.
Histopathological findings reveal diffuse, lobular infiltration by atypical lymphoid cells that appear to circumscribe or “rim” the adipocytes. These findings can mimic those seen in lupus panniculitis, impeding the derivation of a definitive diagnosis. However, features of lupus profundus such as overlying dermal changes consistent with lupus, the presence of lymphoid follicles, lack of clonality, and hyaline sclerosis of the fat can help distinguish these two entities [4]. In SPTL, the neoplastic lymphocytes demonstrate hyperchromatic nuclei, karyorrhexis, and mitoses. Most of the atypical lymphocytes express CD3, CD8, TIA-1 and perforin, but are negative for CD4, CD30. TCR gene analysis by polymerase chain reaction-single strand conformational polymorphism divides SPTL into two subtypes: 1. T cells expressing TCR-α/β and CD8+, and 2. γ/δ T cells often expressing CD56. The most recent WHO-EORTC classification denotes these two different subgroups as SPTL-AB for the former, and SPTL-GD for the latter, though SPTL-GD has been distinguished from SPTL-AB by being placed into a separate, provisional category of γ/δ T cell lymphomas [3]. Even more recently, it was demonstrated that SPTL is a moleculocytogenetically uniform entity of CTCL. SPTL shares chromosome losses of 10q, 17p, and 19 known to be characteristic of mycosis fungoides; it also demonstrates unique chromosome gains of chromosomes 5q and 13 q, distinguishing it from other subtypes of CTCL [7].
The treatment of SPTL is not standardized and may include systemic steroids, cyclosporine, or multi-drug chemotherapy. The prognosis and response is variable depending on systemic symptoms, cytopenia, and associated HPS. The increased proportion of γ/δ subsets and expression of CD56 are more prevalent among patients who develop HPS, suggesting a higher mortality rate among this subset [3,4].
References
1. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Hematopoetic and Lymphoid Tissues. 2001;In Jaffe ES, Harris NL, Stein H, Vardiman JW (Eds.). Lyon France IARC Press.2. Jaffe ES, Harris NL, Stein H, Vardiman JW. WHO classification of tumors. In : Pathology and Genetics of Tumors of Hemopoeitic and Lymphoid tissues. Lyon: IARC Press; 2001.
3. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008; 111: 838-845. [PubMed]
4.Weenig RH, Ng CS, Perniciaro P. Subcutaneous panniculitis-like T-cell lymphoma: An elusive case presenting as lipomembranous panniculitis and a review of 72 cases in the literature. Am J Dermatopathol 2001; 23:206-15. [PubMed]
5. Yung A, Snow J, Jarret P. Subcutaneous panniculitic T-cell lymphoma and cytophagic histiocytic panniculitis. Australas J Dermatol 2001; 42: 183. [PubMed]
6. Craig AJ, Cualing H, Thomas G, Lamerson C, Smith R. Cytophagic histiocytic panniculits: A syndrome associated with benign and malignant panniculits: Case comparison and review of the literature. J Am Acad Dermatol 1998; 39: 721. [PubMed]
7. Hahtola S, Burghart E, Jeskanen L, et al. Clinicopathological characterization and genomic aberrations in subcutaneous panniculitis-like T-cell lymphoma. JID 2008: 128; 2304-9. [PubMed]
© 2010 Dermatology Online Journal